Joint hypermobility is defined as a more-than-normal range of movement (ROM) in a joint (Smits-Engel.dsman 2011). It may occur in a single joint, or in a number off joints (generalised joint hypermobility).
Generalised joint hypermobility is now increasingly recognised as an underlying or contributing factor in movement difficulties experienced by children, both because of the impact of increased compliance in the connective tissue structures of the body but also because it is closely associated with a cautious temperament (behavioural inhibition) and related anxiety.
Terminology
Different terms are used in the literature and by clinicians.
Generalised joint hypermobility refers to the occurrence of hypermobility in multiple joints.
Joint hypermobility syndrome refers to the presence of generalised joint hypermobility associated with pain and altered function.
The term joint hypermobility syndrome is also often used as an abbreviation for Ehlers Danlos Syndrome (Hypermobility Type) .
Recent expert opinion is that clinically JHS and EDD (HT) can be seen as a continuum that there should be no differentiation between the two disorders a a clinical level.
Generalised joint hypermobility
Joint hypermobility is common in childhood, occurring in 8–39% of school age children[. Prevalence depends on age, sex and ethnicity and decreases with increasing age. Girls are generally more hypermobile than boys and children from Asian backgrounds are generally more hypermobile than Caucasian children. (Remvig et al 2007)
The Beighton scale is most commonly used measure for diagnosing GJH. This a nine point scale, measuring range of motion (ROM) at 8 joints plus the ability to put the hands down flat on the floor in standing. Generalised joint hypermobility is defined as scoring 4 or more on the 9 point Beighton scale
The recommended cut-off point for a diagnosis ranges between 5 and 7 for different clinicians and researchers.
In a recent study of 551 children (age range 6-12) attending Dutch primary schools used a standardised protocol to measure the ROM the scores on the Beighton Scale were as follows: 0-4 64%, 5-6 26.5 %&-9 9.1 % Based on these numbers a cut-off point of 7 is recommended (Smits-Engelsman 2001).
9 point Beighton Scale
 Passive dorsiflexion of the fifth metacarpophalangeal joint to < 90 degrees Passive dorsiflexion of the fifth metacarpophalangeal joint to < 90 degrees |
 Passive apposition of the thumb to the flexor side of the forearm, while shoulder is flexed 90 degrees, elbow is extended, and hand is pronated. Passive apposition of the thumb to the flexor side of the forearm, while shoulder is flexed 90 degrees, elbow is extended, and hand is pronated. |
 Passive hyperextension of the elbow < 10 degrees Passive hyperextension of the elbow < 10 degrees |
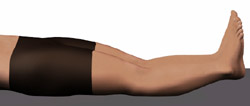 Passive hyperextension of the knee < 10 degrees Passive hyperextension of the knee < 10 degrees |
 Forward flexion of the trunk, with the knees straight, so that the hand palms rest easily on the floor Forward flexion of the trunk, with the knees straight, so that the hand palms rest easily on the floor |
GJH: associated symptoms and disorders
A tendency to having a cautious temperament, scoring high on behavioural inhibition (BI) and fear avoidance items on temperament scales.
Muscle weakness: partly due to the impact of connective tissue compliance on muscle function and partly due to poor engagement with effortful physical activity.
Developmental delay: sitting,standing with support,cruising and walking are often delayed.
There is an increased prevalence of DCD in children with GJH
Proprioception may be poor especially when there is muscle weakness.
Tendency to joint sprains and strains due to joint instability
Gastroesophageal reflux disease (GERD)
Difficulties with bladder and bowl control: related to increased compliance in the bladder and bowel walls as well as sphincter anomalies.
"Patients with generalised joint hypermobility have been shown to be at risk of osteopenia in several studies[17,22-24] so it is worthwhile evaluating bone health in all hypermobile children. Gulbar[23] demonstrated a correlation between increasing Beighton score and decreasing bone density at the hip, measured by DEXA, in a small case-control study of hypermobile adult women after controlling for confounders such as smoking and physical activity. The osteopenia is most likely to be a result of muscle weakness and lack of constraint to joint movement, which decrease the forces transmitted to the bone." (Tofts et al 2009)
Joint Hypermobility Syndrome
Joint hypermobility syndrome (JHS), previously known as benign joint hypermobility syndrome (BJHS), is a heritable disorder of connective tissue that comprises symptomatic hypermobility predisposing to arthralgia, soft tissue injury, and joint instability. It is indistinguishable from the hypermobility type of Ehlers-Danlos syndrome. (Ross 2011)
"There is debate in the literature as to whether isolated joint hypermobility represents the end of the normal spectrum of joint range of movement or whether it represents a polygenic group at the mild end of the spectrum of Heritable Disorders of Connective Tissue[6,7]. It is generally accepted that the phenomenon runs in families and tends to be dominantly inherited. The diagnosis of generalised joint hypermobility, underlying genetic syndromes, and complications such as widespread musculoskeletal pain and chronic fatigue, are largely based on clinical criteria. The genetic causes of joint hypermobility include Ehlers – Danlos syndromes (EDS), some types of Osteogenesis Imperfecta (OI) including types I and IV, Marfan syndrome and related disorders, and rare HDCT such as pseudoxanthoma elasticum and cutis laxa syndromes. Hypermobility may also be a feature of a wide range of skeletal dysplasia syndromes eg pseudoachondroplasia and spondyloepiphyseal dysplasia congenita and developmental syndromes of childhood such as the Fragile-X syndrome." Tofts et al 2009
Disorders and symptoms commonly associated with JHS
Gastrointestinal manifestations include such as chronic constipation, gastro-esophageal reflux, chronic abdominal pain and irritable bowel syndrome
Symptoms related to autonomic dysfunction: syncope, dizziness and orthostatic hypotension and postural tachycardia (Pots)
Mood disorders: Anxiety has been linked to JHS in adults. Clinically children with joint hypermobility often have a very cautious nature.
Chronic pain and fatigue Children with joint hypermobility often experience pain in their joints and muscles. This is expected after injury to a joint but sometimes the pain occurs after even mildly strenuous exercise or without any apparent cause. Read more
Diagnosis
Joint hypermobility syndrome(JHS) is diagnosed when there is joint hypermobility in 4 or more joints along with other signs and symptoms as listed in the Brighton Criteria
Brighton criteria for joint hypermobility syndrome
Major criteria
- Beighton score of ≥4
- Arthralgia for longer than 3 months in four or more joints
Minor criteria
- Beighton score of 1, 2 or 3
- Arthralgia (>3 months) in one to three joints, back pain (>3 months), or spondylosis/spondylolysis/spondylolisthesis
- Dislocation or subluxation in more than one joint, or in one joint on more than one occasion
- Three or more soft tissue lesions (e.g. epicondylitis, tenosynovitis, bursitis)
- Marfanoid habitus (tall, slim, span greater than height (>1.03 ratio), upper segment less than lower segment (<0.89 ratio), arachnodactyly)
- Skin striae, hyperextensibility, thin skin or abnormal scarring
- Ocular signs (e.g. drooping eyelids, myopia, antimongoloid slant)
- Varicose veins, hernia, or uterine or rectal prolapse
- Mitral valve prolapse
Requirements for diagnosis
Any one of the following:
- Two major criteria
- One major criterion plus two minor criteria
- Four minor criteria
- Two minor criteria and an unequivocally affected first-degree relative in family history
Differential diagnosis of joint hypermobility
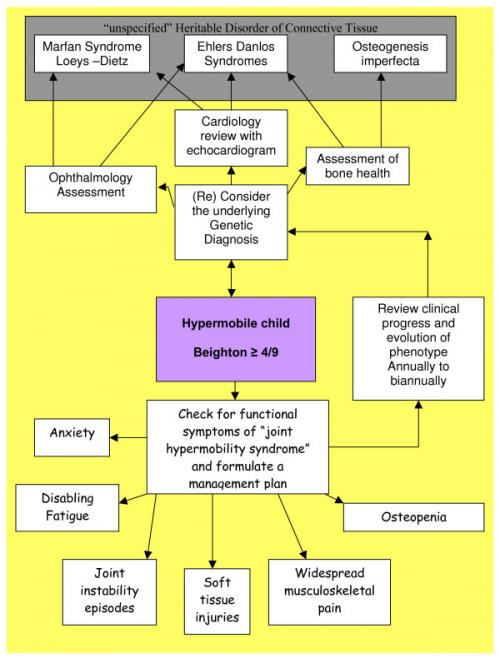
There are a range of connective tissue disorders that have joint hypermobility a a feature. Some of these disorders have particular vascular sometimes life threatening features that need to be recognized. Fot this reason any child with JHS should have a full assessment by a rheumatologist.
The connective tissue dysplasia clinic Children's Hospital at Westmead, Sydney, NSW, Australia) diagnostic approach to a child with hypermobile joints. (Tofts et al 2009)
More about Ehlers Danlos Syndrome
Ehlers-Danlos syndrome (EDS) is a group of inherited disorders marked by extremely loose joints,hyperelastic skin that bruises easily, and easily damaged blood vessels. Because of the possibility of vascular involvement recognizing possible EDS and referring for a dianosis is important.
Ehlers Danlos Classic type
"Ehlers-Danlos syndrome (EDS), classic type is a connective tissue disorder characterized by skin hyperextensibility, abnormal wound healing, and joint hypermobility. It includes two previously designated subtypes (EDS type I and EDS type II) that are now recognized to form a continuum of clinical findings. The skin is smooth, velvety to the touch, and hyperelastic; i.e., it extends easily and snaps back after release (unlike lax, redundant skin, as in cutis laxa). The skin is fragile, as manifested by splitting of the dermis following relatively minor trauma, especially over pressure points (knees, elbows) and areas prone to trauma (shins, forehead, chin). Wound healing is delayed, and stretching of scars after apparently successful primary wound healing is characteristic. Complications of joint hypermobility, such as dislocations of the shoulder, patella, digits, hip, radius, and clavicle, usually resolve spontaneously or are easily managed by the affected individual. Other features include hypotonia with delayed motor development, fatigue and muscle cramps, and easy bruising. Less common findings include mitral and tricuspid valve prolapse, aortic root dilatation, and spontaneous rupture of large arteries." Gene Reviews EDS Classic Type Read more
Ehlers Danlos (Hypermobility Type)
There is a spectrum of generalised joint hypermobility in children and the phenomenon is almost certainly polygenic in origin with environmental influences, particularly participation in sport and flexibility training. Younger children are more flexible but this resolves with increasing age in normal children. It is a clinical challenge to distinguish young children with significant hypermobility who are unlikely to improve from those who are in the normal spectrum of hypermobility and will improve with time. Clinical follow up over several years is currently the only way of answering this question for an individual child.
The current criteria for a diagnosis of Ehlers-Danlos syndrome (hypermobile type) are illustrated in table table22but are non-specific. For a diagnosis to be made an individual needs to have one of the major criteria of a Beighton score of ≥ 4/9 or "Skin involvement (hyperextensibility and/or smooth, velvety skin)". Beighton et al[9] describe testing of skin hyperextensibility at a neutral site e.g. the volar aspect of the forearm where "the skin is pulled up until resistance is felt" but they do not describe a reproducible measurement which can be taken during this test, nor what might be normal and abnormal. Minor criteria which are "of lesser diagnostic specificity" include recurrent joint dislocation, chronic joint/limb pain and a positive family history. The presence of one or more minor criteria is "suggestive" of the diagnosis. The criteria are much clearer than in previous classifications for the more severe subtypes of EDS, but are not specific enough to be very helpful in distinguishing between EDS (hypermobile type) and "normal spectrum" hypermobility. We normally use the diagnosis of Ehlers-Danlos syndrome (hypermobile type) in patients with one major criteria and any one of the minor criteria and tend to diagnose individuals whom we perceive to have very soft skin as EDS (hypermobile type), although we do not have an objective measure of skin quality.
We refer all our hypermobile patients with a Beighton score of ≥ 4/9 for a cardiac and echocardiography assessment to look for mitral valve and aortic root abnormalities. If minor cardiac signs are present we use the EDS (hypermobile type) diagnosis[18] and consider the diagnosis of Marfan syndrome.
Ehlers Danlos Syndrome (Vascular type)
"There are subgroups of children who present with hypermobility and who are at significant risk of mortality as a result of vascular fragility in early adulthood. These children have hypermobility, particularly of small joints, thin translucent skin, lack of subcutaneous fat and bruise easily. They are prone to complications including aortic dissection, stroke from ruptured cerebral vessels and uterine and bowel rupture. There may be a family history of sudden death. EDS (vascular type) is due to a defect in COL3A1 therefore a diagnosis can be made if mutations are found in the type III collagen gene." Tofts et al 2007
Ehlers-Danlos syndrome (EDS), kyphoscoliotic form
"Ehlers-Danlos syndrome (EDS), kyphoscoliotic form (previously known as EDS VI) is a generalized connective tissue disorder characterized by friable, hyperextensible skin, thin scars, and easy bruising; generalized joint laxity; severe muscular hypotonia at birth; progressive scoliosis, present at birth or within the first year of life; and scleral fragility with increased risk of rupture of the globe. Intelligence is normal; life span may be normal, but affected individuals are at risk for rupture of medium-sized arteries and respiratory compromise if kyphoscoliosis is severe." Gene Reviews (EDS), kyphoscoliotic form Read More
Ehlers-Danlos syndrome type IV
Ehlers-Danlos syndrome type IV (EDS type IV) is characterized by thin, translucent skin; easy bruising; characteristic facial appearance (in some individuals); and arterial, intestinal, and/or uterine fragility. Vascular dissection or rupture, gastrointestinal perforation, or organ rupture are the presenting signs in the majority of adults identified to have EDS type IV. Arterial rupture may be preceded by aneurysm, arteriovenous fistulae, or dissection but also may occur spontaneously. Neonates may present with clubfoot and/or congenital dislocation of the hips. In childhood, inguinal hernia, pneumothorax, and recurrent joint subluxation or dislocation can occur. Pregnancy for women with EDS type IV has as much as a 12% risk for death from (EDS type IV)peripartum arterial rupture or uterine rupture. One-fourth of individuals with EDS type IV who have undergone laboratory testing to confirm their diagnosis have experienced a significant medical problem by age 20 years and more than 80% by age 40 years. The median age of death in this reviewed population was 48 years. Gene Review (EDS type IV) Read more
More resources and information for parents
A collection of articles on joint hypermobility and its affect on development and function
Resources
British Society for Paediatric and Adolescent Rheumatology Guidelines for Management of Joint Hypermobility Syndrome in Children and Young People.
References
De Wandele, I., Calders, P., Peersman, W., Rimbaut, S., De Backer, T., Malfait, F., … Rombaut, L. (2014). Autonomic symptom burden in the hypermobility type of Ehlers-Danlos syndrome: a comparative study with two other EDS types, fibromyalgia, and healthy controls. Seminars in Arthritis and Rheumatism, 44(3), 353–61. Link to article
Bale P, Easton V, Bacon H, Jerman E, Watts L, Barton G, Clark A, Armon K, MacGregor AJ. (2019) The effectiveness of a multidisciplinary intervention strategy for the treatment of symptomatic joint hypermobility in childhood: a randomised, single Centre parallel group trial (The Bendy Study). Pediatr Rheumatol Online J. 2019 Jan 8;17(1):2.
Barron, D. F., Cohen, B. a, Geraghty, M. T., Violand, R., & Rowe, P. C. (2002). Joint hypermobility is more common in children with chronic fatigue syndrome than in healthy controls. The Journal of Pediatrics, 141(3), 421–5.
Engelbert RH, Juul-Kristensen B, Pacey V, de Wandele I, Smeenk S, Woinarosky N, Sabo S, Scheper MC, Russek L, Simmonds JV. (2017) The evidence-based rationale for physical therapy treatment of children, adolescents, and adults diagnosed with joint hypermobility syndrome/hypermobile Ehlers Danlos syndrome. Am J Med Genet C Semin Med Genet. 2017 Mar;175(1):158-167. doi: 10.1002/ajmg.c.31545. PubMed PMID: 28306230.
Kovacic, K.,
Chelimsky, T. C., Sood, M. R., Simpson, P., Nugent, M., & Chelimsky, G. (2014). Joint hypermobility: a common association with complex functional gastrointestinal disorders. The Journal of Pediatrics, 165(5), 973–8.
Murray, K. J. (2006). Hypermobility disorders in children and adolescents. Best Practice & Research. Clinical Rheumatology, 20(2), 329–51.
Pacey, V., Adams, R. D., Tofts, L., Munns, C. F., & Nicholson, L. L. (2014). Proprioceptive acuity into knee hypermobile range in children with joint hypermobility syndrome. Pediatric Rheumatology Online Journal, 12, 40. doi:10.1186/1546-0096-12-40
Pacey, V., Adams, R. D., Tofts, L., Munns, C. F., & Nicholson, L. L. (2015). Joint hypermobility syndrome subclassification in paediatrics: a factor analytic approach. Archives of Disease in Childhood, 100(1), 8–13. doi:10.1136/archdischild-2013-305304 Link to article
Pacey, V., Tofts, L., Wesley, A., Collins, F., & Singh-Grewal, D. (2014). Joint hypermobility syndrome: A review for clinicians. Journal of Paediatrics and Child Health, 1–8. doi:10.1111/jpc.12731
Pacey V, Tofts L, Adams RD, Munns CF, Nicholson LL.(2013) Exercise in children with joint hypermobility syndrome and knee pain: a randomised controlled trial comparing exercise into hypermobile versus neutral knee extension. Pediatr Rheumatol Online J. 2013 Aug 14;11(1):30. doi: 10.1186/1546-0096-11-30.
Plackett, T. P., Kwon, E.,
Gagliano, R. a, & Oh, R. C. (2014). Ehlers-danlos syndrome-hypermobility type and hemorrhoids. Case Reports in Surgery, 2014, Link to article
Remvig L, Jensen DV, Ward RC. Epidemiology of general joint hypermobility and basis for the proposed criteria for benign joint hypermobility syndrome: review of the literature. J Rheumatol.2007;34:804–809. Request via Research Gate
Rombaut, L., Scheper, M., De Wandele, I., De Vries, J., Meeus, M., Malfait, F., … Calders, P. (2014). Chronic pain in patients with the hypermobility type of Ehlers-Danlos syndrome: evidence for generalized hyperalgesia. Clinical Rheumatology. doi:10.1007/s10067-014-2499-0 Link to article
Shirley, E. D., Demaio, M., & Bodurtha, J. (2012). Ehlers-danlos syndrome in orthopaedics: etiology, diagnosis, and treatment implications. Sports Health, 4(5), 394–403. doi:10.1177/1941738112452385
Smits-Engelsman, B., Klerks, M., & Kirby, A. (2011). Beighton score: a valid measure for generalized hypermobility in children. The Journal of Pediatrics, 158(1), 119–23, 123.e1–4. doi:10.1016/j.jpeds.2010.07.021 PDF
Tofts, L. J., Elliott, E. J., Munns, C., Pacey, V., & Sillence, D. O. (2009). The differential diagnosis of children with joint hypermobility: a review of the literature. Pediatric Rheumatology Online Journal, 7, 1. doi:10.1186/1546-0096-7-1 Link to article
Voermans, N. C., Knoop, H., van de Kamp, N., Hamel, B. C., Bleijenberg, G., & van Engelen, B. G. (2010). Fatigue is a frequent and clinically relevant problem in Ehlers-Danlos Syndrome. Seminars in Arthritis and Rheumatism, 40(3), 267–74. doi:10.1016/j.semarthrit.2009.08.003 Request via Research Gate

